Персональный сайт пользователя
Тави Тум
:
bosnn .www.nn.ru
пользователь имеет статус «трастовый»
портрет №
64828 зарегистрирован
в 2007 году
Тави Тум
она же
Ю нона
по
26-03-2011
настоящее имя:
Занятия для детей с нарушением памяти и внимания
(
Подготовка к школе )

Портрет заполнен на
78 %
Отправить приватное сообщение Добавить в друзья Игнорировать Сделать подарок
Уровни
Тави Тум на других форумах

- сейчас просматривают портрет - 0
- зарегистрированные пользователи посетившие портрет за 7 дней - 2
Блог
>
Георгий Базыкин " Протокол эпидемии: что может сказать биоинформатика о вспышке nCoV"
14.02.2020 в 20:49 424

Просмотреть или сохранить оригинал: Георгий Базыкин " Протокол эпидеми...
Георгий Базыкин - кандидат биологических наук, PhD Принстонского университета, зав. сектором молекулярной эволюции ИППИ РАН, ведущий научный сотрудник Факультета биоинженерии и биоинформатики МГУ
Мы находимся в разгаре эпидемии новой опасной инфекции, но достоверной информации о ней мало. В этой ситуации помогает молекулярная эпидемиология. Ход накопления мутаций эволюционирующим вирусом позволяет оценить важные параметры эпидемии даже при неточных официальных данных. Ситуация очень быстро меняется; все оценки, приведенные ниже, верны по состоянию на 9 февраля.
С чего все началось?
Сравнивая организмы друг с другом, эволюционные биологи многое могут узнать про их историю. Больше всего материала для сравнений содержится в нуклеотидных последовательностях, в случае коронавирусов — в последовательностях РНК. Текущая вспышка — пожалуй, первый случай возникновения нового патогена, когда генетические данные есть почти с самого начала. Первая последовательность «нового» коронавируса (nCoV) появилась в открытом доступе всего через несколько недель после описания первых случаев [1]; для сравнения: при вспышке SARS (тяжелого острого респираторного синдрома, также вызываемого коронавирусами) 2002/2003 года первых последовательностей пришлось ждать много месяцев.
Рис. 1. Эволюционное древо коронавирусов (из работы [4]). Числа около ветвей обозначают их статистическую достоверность по шкале от 0 до 100. Красным выделены штаммы уханьской вспышки (Wuhan), а также ближайший к ним штамм, выделенный из летучей мыши (Bat CoV RaTG13)
Рис. 1. Эволюционное древо коронавирусов (из работы [4]). Числа около ветвей обозначают их статистическую достоверность по шкале от 0 до 100. Красным выделены штаммы уханьской вспышки (Wuhan), а также ближайший к ним штамм, выделенный из летучей мыши (Bat CoV RaTG13)
На рис. 1 показано эволюционное дерево коронавирусов. Как и на обычных эволюционных деревьях, расстояние между любыми двумя ветвями здесь соответствует времени, прошедшему с момента расхождения двух линий от их общего предка (LCA — last common ancestor). Видно, что ближайший родственник группы вирусных штаммов, давших начало этой вспышке, — это коронавирус, выделенный из летучей мыши; между ним и эпидемическими штаммами совпадает около 96% нуклеотидов. Такая картина может означать, что вирус был передан от летучих мышей, хотя окончательные выводы о пути передачи делать рано; в случае SARS, чтобы разобраться, потребовались годы. (Сведения о еще более близких штаммах из панголина пока есть только в виде пресс-релиза [2]). В любом случае все имеющиеся данные свидетельствуют о том, что инфекция была получена из природного резервуара. Чуть дальше расположены штаммы SARS, с которыми совпадает ~80% нуклеотидов; а еще дальше — штаммы MERS, ближневосточного респираторного синдрома [3, 4]. В последовательностях nCoV нет никаких свидетельств искусственной рекомбинации, вставок необычных фрагментов или какого-либо вмешательства; все сообщения об обратном, появлявшиеся в последние дни на препринтных серверах, были отозваны и/или опровергнуты.
Все линии nCoV более близки друг к другу, чем к любой другой известной вирусной последовательности. Это, по-видимому, означает, что занос был единственным. Не всегда бывает так: например, вспышки MERS в разные годы вызывались новыми переносами из природного резервуара — верблюдов [5].
Зная скорость эволюции (см. ниже), можно датировать LCA. По-видимому, он существовал в конце ноября — начала декабря. LCA мог быть у единственного человека, который заразил потом других, или же у животного, от которого заразилось несколько человек — это установить трудно. Первые описанные случаи датируются началом декабря; это означает, что вспышка была обнаружена почти сразу. Образцы вируса, выделенные с рынка в Ухане, очень близки к таковым у самых ранних пациентов из Уханя; это подтверждает, что первые люди заразились именно там.
Неизвестно, как мутации, отличающие nCoV от штаммов, распространенных у животных, изменили его характеристики и изменили ли вообще. Возможно, перескок был несчастной случайностью, не сопровождавшейся никакими изменениями генетических характеристик вируса по сравнению с его предками у диких животных.
Что происходит сейчас?
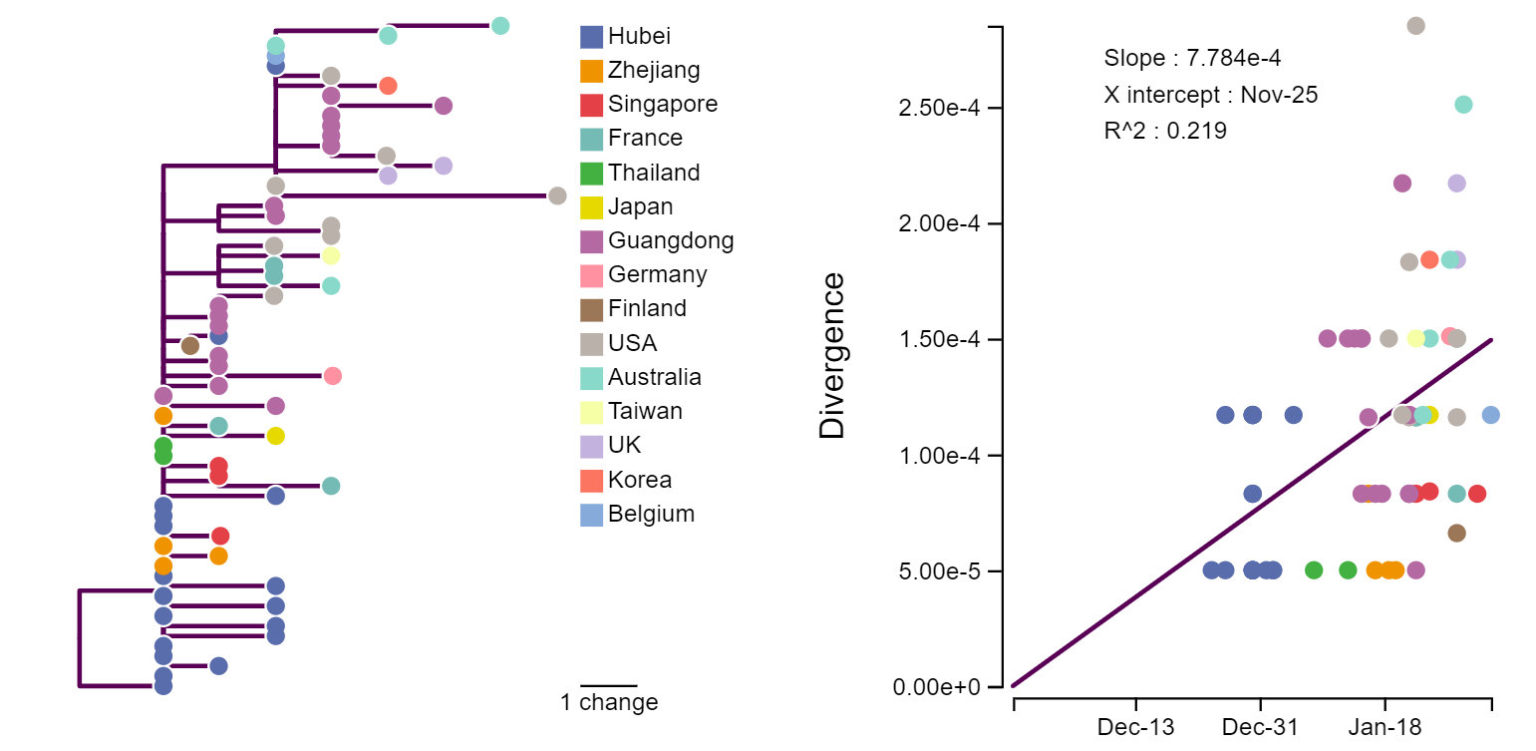
Любые биологические объекты изменяются в ряду поколений из-за случайных мутаций. «Перескочив» в людей, вирус продолжил эволюционировать, «прорастив» ветвистое эволюционное древо уже в людях (рис. 2) К сожалению, сегодня известны лишь очень немногие «листья» этого древа. Самые новые последовательности из Уханя датируются 3 января, и 42 из 73 известных сегодня последовательностей получены снаружи Китая (при том, что 99% подтвержденных случаев — в Китае [6]). Тем не менее, изучая древо, можно понять довольно много.
Рис. 2. Эволюционное древо nCoV [7] (слева) и расстояние между образцом и корнем дерева в зависимости от даты получения образца
Рис. 2. Эволюционное древо nCoV [7] (слева) и расстояние между образцом и корнем дерева в зависимости от даты получения образца
Во-первых, можно установить скорость эволюции вируса. Для этого нужно сопоставить даты инфицирования с числом отличий последовательности вируса от «предковой». По текущим оценкам, скорость составляет порядка 10–3 замен на нуклеотид в год [7, 8]; это сопоставимо с таковой у других РНК-содержащих вирусов, например, у гриппа [9]. Это может означать, что, как и в случае с гриппом, трудно будет создать универсальную вакцину, защищающую от всех штаммов, и вакцина потребует периодического обновления.
Во-вторых, можно проследить пути передачи. Первые надежные данные о передаче от человека человеку были получены именно таким образом. Можно задавать и более тонкие вопросы. Сколько людей заражается от членов семьи, сколько на работе, сколько в транспорте, сколько в больнице? Насколько эффективны карантинные меры? Как именно вирус переносится между странами? Можно пытаться узнать это по молекулярным данным. Это очень помогает с другими инфекциями, для которых таких данных больше, например с ВИЧ [10].
В-третьих, можно оценить скорость распространения вируса, а именно ставший знаменитым в последние дни показатель R0. R0, или базовое репродуктивное число, — это число людей, зараженных одним инфицированным за всё время течения болезни в полностью уязвимой популяции. Очевидно, что это ключевой показатель: если R0 меньше единицы, то эпидемия пойдет на спад, а если больше — то будет расти. Разные заболевания характеризуются очень разными значениями R0 — от 1,3 для гриппа до >10 для кори. Для ближайшего родственника nCoV — SARS — R0 составлял около 3 в начале вспышки и около 0,3 в ее конце [11].
Как измерить R0? Можно строить «традиционные» эпидемические модели и оценивать скорость роста числа заболевших. К сожалению, это трудно сделать точно. Начало вспышки трудно датировать, а в разгар эпидемии многие легкие случаи заведомо не диагностируются; даже скорость диагностики тяжелых случаев может зависеть от пропускной способности системы здравоохранения, например от доступности тест-систем.
Эволюционные методы дают другие способы оценить скорость распространения эпидемии. Основная идея такова: общие статистические характеристики древа, например отношения длин ветвей вблизи корня к длинам ветвей около листьев, должны зависеть от того, сохраняет ли патоген приблизительно постоянную численность или же она растет или убывает. Это связано с тем, что длина ветвей древа, построенного по выборке из некой популяции, определяется численностью этой популяции: чем меньше популяция, тем быстрее «встречаются» друг с другом ветви, поскольку повышается вероятность, что две случайно выбранные особи оказываются близкими родственниками. Растущая вспышка вируса определяется относительно короткими ветвями у корня и относительно длинными — около листьев.
Применение такого анализа к nCoV дает оценки R0 в районе 2 или 3 [7, 8]). Конечно, здесь тоже много предположений, многие из которых заведомо неверны: например, что выборка анализируемых последовательностей более-менее случайна и что на вирус не действует отбор. Но данные, полученные с использованием разных методов, взаимно подтверждают друг друга и позволяют сказать, что R0, по-видимому, находится где-то в диапазоне от 2,2 до 3,3 [12].
Что может быть дальше?
В последние дни появились работы, в которых моделируется развитие эпидемии при различных параметрах [13, 14, 15]. Важно понимать, что эти модели — не прогнозы. Во-первых, точность, с которой оценен R0, совершенно недостаточна. При R0~3 в отсутствие каких-либо мер и без предсуществующего иммунитета вспышка, например, в десятимиллионном городе будет быстрой и острой, будет иметь пик через два-три месяца, и на пике будут одновременно инфицированы десятки процентов населения [15]. Если же R0 ниже двух, то пик растянется на много месяцев и будет смазан (рис. 3).
Рис. 3. Число инфицированных в простейшей детерминистической SIR-модели [16]. Параметры: N=107, γ=0,119
Рис. 3. Число инфицированных в простейшей детерминистической SIR-модели [16]. Параметры: N=107, γ=0,119
Во-вторых, практически невозможно моделировать такие факторы, как разработка вакцин, эффективность имеющихся лекарственных препаратов (которая остается пока практически неизвестной), а также принимаемые меры по снижению скорости распространения.
В-третьих, сам по себе R0 мало что говорит о том, насколько серьезной окажется эпидемия: R0 для риновирусов, вызывающих обыкновенную простуду, составляет ~6, однако они не являются столь существенной проблемой мирового здравоохранения. Ключевые параметры, остающиеся неизвестными, — это доля тяжелых и смертельных случаев. Составит ли она ~0,1% от общего числа инфицированных, как для ежегодного сезонного гриппа, 2,5%, как для испанки, или 10%, как для SARS? Эти сценарии будут очень разными. Имеющиеся сегодня данные не позволяют сказать, какой из них сбудется.
Георгий Базыкин
(Сколтех, ИППИ РАН)
Wuhan seafood market pneumonia virus isolate Wuhan-Hu-1, complete genome. (2020).
Cyranoski D. Did pangolins spread the China coronavirus to people? Nature (2020) doi: 10.1038/d41586-020-00364-2.
Lu R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet 10 (2020).
Zhou P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 1–4 (2020) doi: 10.1038/s41586-020-2012-7.
Zhang Z., Shen L. & Gu X. Evolutionary Dynamics of MERS-CoV: Potential Recombination, Positive Selection and Transmission. Sci Rep 6, (2016).
Coronavirus 2019-nCoV global cases by Johns Hopkins CSSE.
Rambaut A. Phylodynamic Analysis | 67 genomes | 08 Feb 2020. Virological (2020).
Bedford T. Nextstrain / narratives / ncov / sit-rep / 2020-01-30 (2020).
Peck K. M. & Lauring, A. S. Complexities of Viral Mutation Rates. Journal of Virology 92, (2018).
Poon A. F. Y. et al. Near real-time monitoring of HIV transmission hotspots from routine HIV genotyping: an implementation case study. Lancet HIV 3, e231-238 (2016).
World Health Organization. Consensus document on the epidemiology of severe acute respiratory syndrome (SARS). (2003).
ncov-R0. Google Docs.
Read J. M., Bridgen J. R., Cummings D. A., Ho A. & Jewell C. P. Novel coronavirus 2019-nCoV: early estimation of epidemiological parameters and epidemic predictions. medRxiv 2020.01.23.20018549 (2020) doi: 10.1101/2020.01.23.20018549.
Riou J. & Althaus C. L. Pattern of early human-to-human transmission of Wuhan 2019-nCoV. bioRxiv 2020.01.23.917351 (2020) doi: 10.1101/2020.01.23.917351.
Wu J. T., Leung K. & Leung, G. M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. The Lancet (2020) doi: 10.1016/S0140-6736(20)30260-9.
Compartmental models in epidemiology. Wikipedia (2020).
Мы находимся в разгаре эпидемии новой опасной инфекции, но достоверной информации о ней мало. В этой ситуации помогает молекулярная эпидемиология. Ход накопления мутаций эволюционирующим вирусом позволяет оценить важные параметры эпидемии даже при неточных официальных данных. Ситуация очень быстро меняется; все оценки, приведенные ниже, верны по состоянию на 9 февраля.
С чего все началось?
Сравнивая организмы друг с другом, эволюционные биологи многое могут узнать про их историю. Больше всего материала для сравнений содержится в нуклеотидных последовательностях, в случае коронавирусов — в последовательностях РНК. Текущая вспышка — пожалуй, первый случай возникновения нового патогена, когда генетические данные есть почти с самого начала. Первая последовательность «нового» коронавируса (nCoV) появилась в открытом доступе всего через несколько недель после описания первых случаев [1]; для сравнения: при вспышке SARS (тяжелого острого респираторного синдрома, также вызываемого коронавирусами) 2002/2003 года первых последовательностей пришлось ждать много месяцев.
Рис. 1. Эволюционное древо коронавирусов (из работы [4]). Числа около ветвей обозначают их статистическую достоверность по шкале от 0 до 100. Красным выделены штаммы уханьской вспышки (Wuhan), а также ближайший к ним штамм, выделенный из летучей мыши (Bat CoV RaTG13)
Рис. 1. Эволюционное древо коронавирусов (из работы [4]). Числа около ветвей обозначают их статистическую достоверность по шкале от 0 до 100. Красным выделены штаммы уханьской вспышки (Wuhan), а также ближайший к ним штамм, выделенный из летучей мыши (Bat CoV RaTG13)
На рис. 1 показано эволюционное дерево коронавирусов. Как и на обычных эволюционных деревьях, расстояние между любыми двумя ветвями здесь соответствует времени, прошедшему с момента расхождения двух линий от их общего предка (LCA — last common ancestor). Видно, что ближайший родственник группы вирусных штаммов, давших начало этой вспышке, — это коронавирус, выделенный из летучей мыши; между ним и эпидемическими штаммами совпадает около 96% нуклеотидов. Такая картина может означать, что вирус был передан от летучих мышей, хотя окончательные выводы о пути передачи делать рано; в случае SARS, чтобы разобраться, потребовались годы. (Сведения о еще более близких штаммах из панголина пока есть только в виде пресс-релиза [2]). В любом случае все имеющиеся данные свидетельствуют о том, что инфекция была получена из природного резервуара. Чуть дальше расположены штаммы SARS, с которыми совпадает ~80% нуклеотидов; а еще дальше — штаммы MERS, ближневосточного респираторного синдрома [3, 4]. В последовательностях nCoV нет никаких свидетельств искусственной рекомбинации, вставок необычных фрагментов или какого-либо вмешательства; все сообщения об обратном, появлявшиеся в последние дни на препринтных серверах, были отозваны и/или опровергнуты.
Все линии nCoV более близки друг к другу, чем к любой другой известной вирусной последовательности. Это, по-видимому, означает, что занос был единственным. Не всегда бывает так: например, вспышки MERS в разные годы вызывались новыми переносами из природного резервуара — верблюдов [5].
Зная скорость эволюции (см. ниже), можно датировать LCA. По-видимому, он существовал в конце ноября — начала декабря. LCA мог быть у единственного человека, который заразил потом других, или же у животного, от которого заразилось несколько человек — это установить трудно. Первые описанные случаи датируются началом декабря; это означает, что вспышка была обнаружена почти сразу. Образцы вируса, выделенные с рынка в Ухане, очень близки к таковым у самых ранних пациентов из Уханя; это подтверждает, что первые люди заразились именно там.
Неизвестно, как мутации, отличающие nCoV от штаммов, распространенных у животных, изменили его характеристики и изменили ли вообще. Возможно, перескок был несчастной случайностью, не сопровождавшейся никакими изменениями генетических характеристик вируса по сравнению с его предками у диких животных.
Что происходит сейчас?
Любые биологические объекты изменяются в ряду поколений из-за случайных мутаций. «Перескочив» в людей, вирус продолжил эволюционировать, «прорастив» ветвистое эволюционное древо уже в людях (рис. 2) К сожалению, сегодня известны лишь очень немногие «листья» этого древа. Самые новые последовательности из Уханя датируются 3 января, и 42 из 73 известных сегодня последовательностей получены снаружи Китая (при том, что 99% подтвержденных случаев — в Китае [6]). Тем не менее, изучая древо, можно понять довольно много.
Рис. 2. Эволюционное древо nCoV [7] (слева) и расстояние между образцом и корнем дерева в зависимости от даты получения образца
Рис. 2. Эволюционное древо nCoV [7] (слева) и расстояние между образцом и корнем дерева в зависимости от даты получения образца
Во-первых, можно установить скорость эволюции вируса. Для этого нужно сопоставить даты инфицирования с числом отличий последовательности вируса от «предковой». По текущим оценкам, скорость составляет порядка 10–3 замен на нуклеотид в год [7, 8]; это сопоставимо с таковой у других РНК-содержащих вирусов, например, у гриппа [9]. Это может означать, что, как и в случае с гриппом, трудно будет создать универсальную вакцину, защищающую от всех штаммов, и вакцина потребует периодического обновления.
Во-вторых, можно проследить пути передачи. Первые надежные данные о передаче от человека человеку были получены именно таким образом. Можно задавать и более тонкие вопросы. Сколько людей заражается от членов семьи, сколько на работе, сколько в транспорте, сколько в больнице? Насколько эффективны карантинные меры? Как именно вирус переносится между странами? Можно пытаться узнать это по молекулярным данным. Это очень помогает с другими инфекциями, для которых таких данных больше, например с ВИЧ [10].
В-третьих, можно оценить скорость распространения вируса, а именно ставший знаменитым в последние дни показатель R0. R0, или базовое репродуктивное число, — это число людей, зараженных одним инфицированным за всё время течения болезни в полностью уязвимой популяции. Очевидно, что это ключевой показатель: если R0 меньше единицы, то эпидемия пойдет на спад, а если больше — то будет расти. Разные заболевания характеризуются очень разными значениями R0 — от 1,3 для гриппа до >10 для кори. Для ближайшего родственника nCoV — SARS — R0 составлял около 3 в начале вспышки и около 0,3 в ее конце [11].
Как измерить R0? Можно строить «традиционные» эпидемические модели и оценивать скорость роста числа заболевших. К сожалению, это трудно сделать точно. Начало вспышки трудно датировать, а в разгар эпидемии многие легкие случаи заведомо не диагностируются; даже скорость диагностики тяжелых случаев может зависеть от пропускной способности системы здравоохранения, например от доступности тест-систем.
Эволюционные методы дают другие способы оценить скорость распространения эпидемии. Основная идея такова: общие статистические характеристики древа, например отношения длин ветвей вблизи корня к длинам ветвей около листьев, должны зависеть от того, сохраняет ли патоген приблизительно постоянную численность или же она растет или убывает. Это связано с тем, что длина ветвей древа, построенного по выборке из некой популяции, определяется численностью этой популяции: чем меньше популяция, тем быстрее «встречаются» друг с другом ветви, поскольку повышается вероятность, что две случайно выбранные особи оказываются близкими родственниками. Растущая вспышка вируса определяется относительно короткими ветвями у корня и относительно длинными — около листьев.
Применение такого анализа к nCoV дает оценки R0 в районе 2 или 3 [7, 8]). Конечно, здесь тоже много предположений, многие из которых заведомо неверны: например, что выборка анализируемых последовательностей более-менее случайна и что на вирус не действует отбор. Но данные, полученные с использованием разных методов, взаимно подтверждают друг друга и позволяют сказать, что R0, по-видимому, находится где-то в диапазоне от 2,2 до 3,3 [12].
Что может быть дальше?
В последние дни появились работы, в которых моделируется развитие эпидемии при различных параметрах [13, 14, 15]. Важно понимать, что эти модели — не прогнозы. Во-первых, точность, с которой оценен R0, совершенно недостаточна. При R0~3 в отсутствие каких-либо мер и без предсуществующего иммунитета вспышка, например, в десятимиллионном городе будет быстрой и острой, будет иметь пик через два-три месяца, и на пике будут одновременно инфицированы десятки процентов населения [15]. Если же R0 ниже двух, то пик растянется на много месяцев и будет смазан (рис. 3).
Рис. 3. Число инфицированных в простейшей детерминистической SIR-модели [16]. Параметры: N=107, γ=0,119
Рис. 3. Число инфицированных в простейшей детерминистической SIR-модели [16]. Параметры: N=107, γ=0,119
Во-вторых, практически невозможно моделировать такие факторы, как разработка вакцин, эффективность имеющихся лекарственных препаратов (которая остается пока практически неизвестной), а также принимаемые меры по снижению скорости распространения.
В-третьих, сам по себе R0 мало что говорит о том, насколько серьезной окажется эпидемия: R0 для риновирусов, вызывающих обыкновенную простуду, составляет ~6, однако они не являются столь существенной проблемой мирового здравоохранения. Ключевые параметры, остающиеся неизвестными, — это доля тяжелых и смертельных случаев. Составит ли она ~0,1% от общего числа инфицированных, как для ежегодного сезонного гриппа, 2,5%, как для испанки, или 10%, как для SARS? Эти сценарии будут очень разными. Имеющиеся сегодня данные не позволяют сказать, какой из них сбудется.
Георгий Базыкин
(Сколтех, ИППИ РАН)
Wuhan seafood market pneumonia virus isolate Wuhan-Hu-1, complete genome. (2020).
Cyranoski D. Did pangolins spread the China coronavirus to people? Nature (2020) doi: 10.1038/d41586-020-00364-2.
Lu R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet 10 (2020).
Zhou P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 1–4 (2020) doi: 10.1038/s41586-020-2012-7.
Zhang Z., Shen L. & Gu X. Evolutionary Dynamics of MERS-CoV: Potential Recombination, Positive Selection and Transmission. Sci Rep 6, (2016).
Coronavirus 2019-nCoV global cases by Johns Hopkins CSSE.
Rambaut A. Phylodynamic Analysis | 67 genomes | 08 Feb 2020. Virological (2020).
Bedford T. Nextstrain / narratives / ncov / sit-rep / 2020-01-30 (2020).
Peck K. M. & Lauring, A. S. Complexities of Viral Mutation Rates. Journal of Virology 92, (2018).
Poon A. F. Y. et al. Near real-time monitoring of HIV transmission hotspots from routine HIV genotyping: an implementation case study. Lancet HIV 3, e231-238 (2016).
World Health Organization. Consensus document on the epidemiology of severe acute respiratory syndrome (SARS). (2003).
ncov-R0. Google Docs.
Read J. M., Bridgen J. R., Cummings D. A., Ho A. & Jewell C. P. Novel coronavirus 2019-nCoV: early estimation of epidemiological parameters and epidemic predictions. medRxiv 2020.01.23.20018549 (2020) doi: 10.1101/2020.01.23.20018549.
Riou J. & Althaus C. L. Pattern of early human-to-human transmission of Wuhan 2019-nCoV. bioRxiv 2020.01.23.917351 (2020) doi: 10.1101/2020.01.23.917351.
Wu J. T., Leung K. & Leung, G. M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. The Lancet (2020) doi: 10.1016/S0140-6736(20)30260-9.
Compartmental models in epidemiology. Wikipedia (2020).